Protein research is one of the most challenging topics in analytical chemistry. Initial protein research focused on developing techniques capable of isolating and identifying major sequences (or portions) of proteins. The Edman degradation of peptides is a revolutionary method to identify the main amino acid sequence of a peptide. In addition, the whole sequence of proteins can be determined by combining protein digestion with different enzymes, fractionation of peptides by high performance liquid chromatography (HPLC), and then Edman degradation. In the 1990s, protein identification by Edman degradation was replaced by mass spectrometry due to its ease of operation and ability to achieve results. The study of protein collections in samples was also made possible. Over the years, more techniques are developed for studying proteins, an area that becomes so popular that the term “proteomics” is designated for it. Proteomics was used to define large-scale studies of the proteome, but now refers to small-scale to large-scale protein studies.
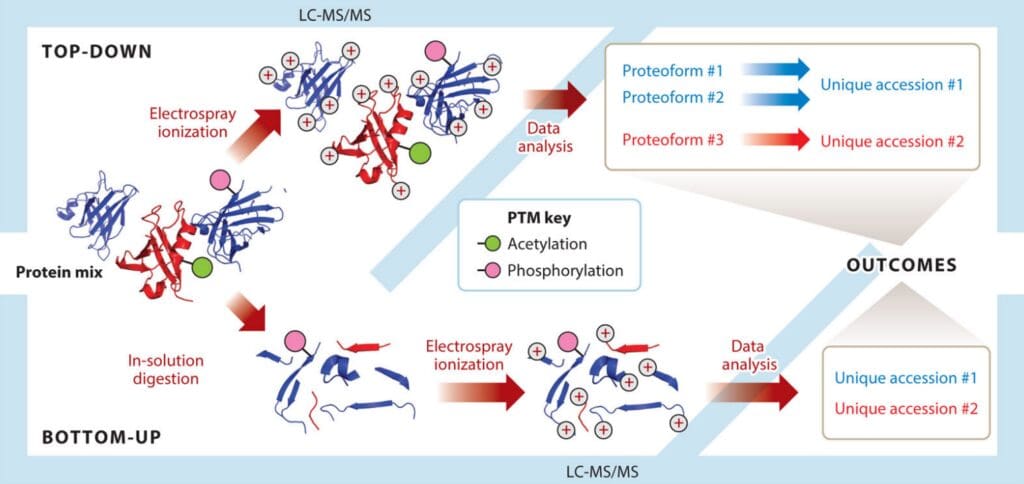
Bottom-up and top-down are two biomass spectrometry-based methods to analyze proteomics. Bottom-up, also known as shotgun, is a widely used mass spectrometry technique in proteomic research, a traditional method that digests/enzymatically dissolves large protein fragments into small peptides for analysis. Top-down can directly sequence intact proteins, including post-translational modified proteins and other large fragment proteins, rather than just peptides.
Bottom-Up Proteomics
The basic process is that the protein mixture is digested into a peptide mixture with or without separation. After the peptide mixture is chromatographed and ionized, the peptide fragment fingerprints are generated by tandem mass spectrometry for peptide identification. Finally, possible proteins were deduced from the identified peptides. This method can obtain a large number of identification results in a short time.
In the calculation step, the existing analytical map methods include a sequence library search, a map library search (most used), de novo sequencing, and a method of de novo sequencing combined with fault tolerance search. Some classic library search software has been widely used, including MASCOT, SEQUEST, X! Tandem and so on.
Taking the shotgun technology route as an example, the basic process of the sequence library search method is as follows:
a.Theoretically cut the candidate protein sequence in the database into peptides, and simulate the fragmentation map of the theoretically cut peptides.
b.The experimental maps were matched and scored based on the similarity of the maps. The highly reliable peptide identification results were obtained by specific peptide quality control methods.
c.Protein was derived based on the correspondence between the peptides and the amino acid sequence of the protein.
Currently, bottom-up strategy is the most mature and widely used to identify proteins, characterize post-translational modifications, and perform relative and absolute quantification. At the same time, however, internal flaws in this strategy limit its potential application.
Top-Down Proteomics
Although many protein separation techniques have been developed, most do not have the ability to single out a protein from a complex mixture. A top-down proteomics strategy has been developed to characterize multiple intact proteins in a mixture. In top-down proteomics, intact proteins are first separated from complex biological samples by reversed-phase liquid chromatography, and then ionized directly by electrospray ionization (ESI) or matrix-assisted laser desorption/ionization (MALDI) technology. The generated ions are fragmented by collision induced dissociation (CID), high energy collision induced dissociation (HCD), electron capture dissociation (ECD) or electron transfer dissociation (ETD), and analyzed in tandem mass spectrometry. This method is promising for protein identification, analysis, sequence analysis, and post-translational modification characterization.
Both the top-down and bottom-up strategies have their advantages and limitations. Considering the complementarity of the information provided by the two strategies, a “middle-down” proteomics strategy is gradually derived, in which large proteins are subject to limited proteolysis by enzymes such as LysC, producing products in the 5–20 kDa range. These peptides are then sequenced using a top-down approach, which has the advantage of high sequence coverage and retention of PTM information. It is important to choose the appropriate protein research strategy according to the equipment and specific analysis requirements.
References
1. Zhou H, Ning Z, E. Starr A, et al. Advancements in top-down proteomics. Analytical chemistry, 2012, 84(2): 720-734.
2. Fornelli L, Toby T K, Schachner L F, et al. Top-down proteomics: where we are, where we are going? Journal of proteomics, 2018, 175: 3.
3. Zhang Y, Fonslow B R, Shan B, et al. Protein analysis by shotgun/bottom-up proteomics. Chemical reviews, 2013, 113(4): 2343-2394.
4. Di Meo A, Pasic M D, Yousef G M. Proteomics and peptidomics: moving toward precision medicine in urological malignancies. Oncotarget, 2016, 7(32): 52460.
5. Visit our website.



